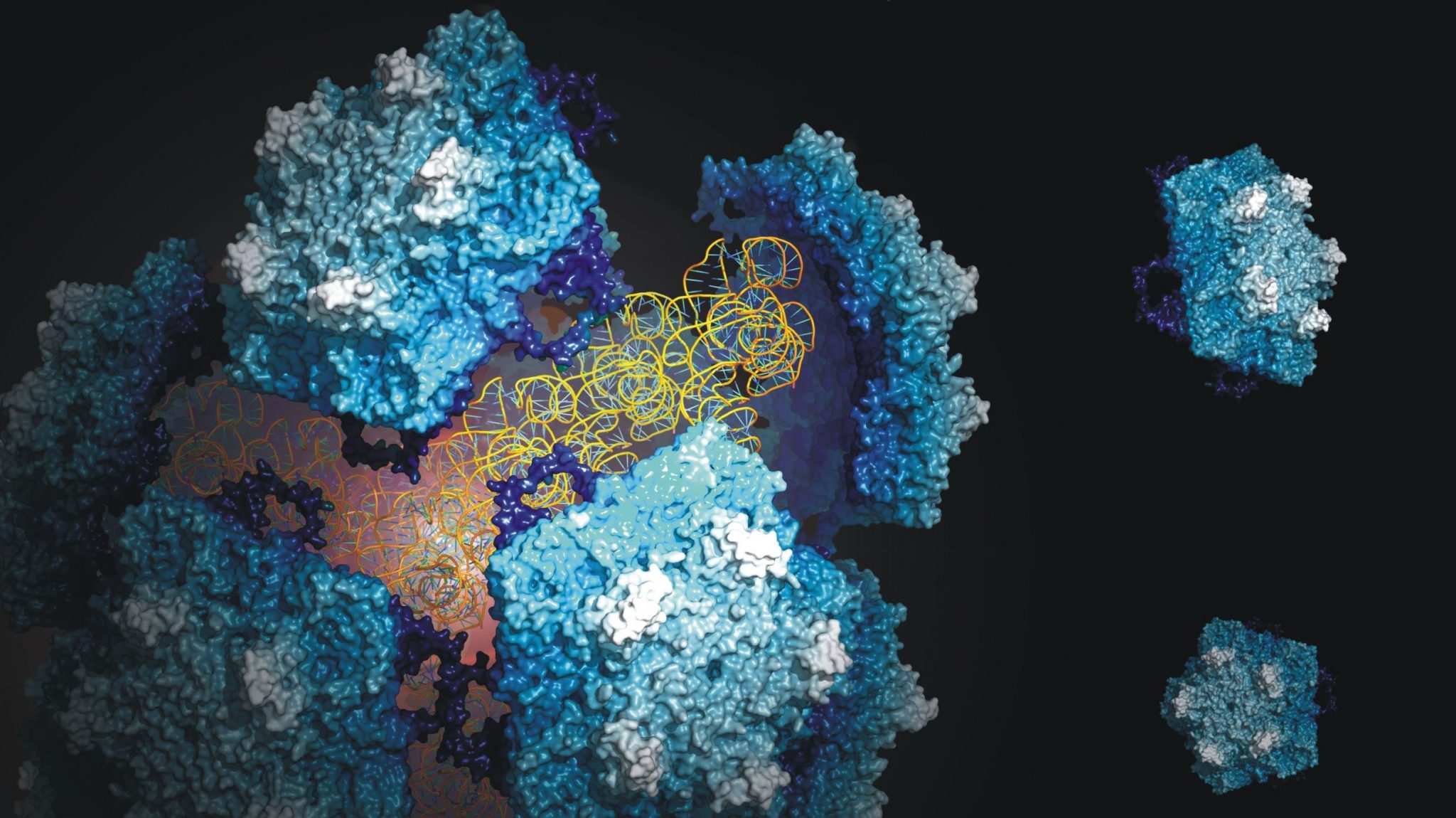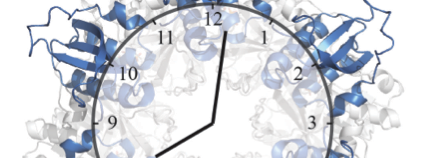

Almost all living things have adapted to earth’s day and night rhythm by keeping time with a biological clock. The biological clock of cyanobacteria, known as the Kai system, is one of the simplest known to date, and is therefore intensively studied by theoretical biologists and systems biologists in the hope to learn about biological time keeping. The Kai system is so robust, that it can even be reconstituted in the lab, simply by mixing the three Kai proteins (KaiA, KaiB and KaiC) in a test-tube. This test-tube oscillator still produces the same 24 hour rhythm that it does in the context of a living cell, and it can do so for weeks on end. The three Kai proteins interact with each other to produce these 24 hour rhythms, forming large protein complexes whose structure and composition changes depending on the time of day. The interactions between KaiC and KaiB marks a defining moment in the 24 hour cycle of the clock, but structural details of this interaction have not been well understood.
In the Proceedings of the National Academy of Sciences USA, Joost Snijder, Rebecca Burnley and Albert Heck, from the Biomolecular Mass Spectrometry and Proteomics Group (Utrecht University), have used structural mass spectrometry (native MS, ion mobility MS and HDX-MS) to shed light on the interaction between KaiB and KaiC. Their experiments revealed the composition of the KaiC-KaiB complex and showed which regions of the proteins are important for their interaction. Understanding these aspects of the KaiB-KaiC interaction explains many of the known features of the biological clock and helps us understand how such a simple system by biological standards is able to constitute a complex time keeping device in cyanobacteria.
The work was performed in collaboration with the groups of Alexandre Bonvin (Computational Structural Biology, Bijvoet Centre, Utrecht University) and Ilka Axmann (Institute for Theoretical Biology, Universitätsmedizin Berlin)
Link to the full article: