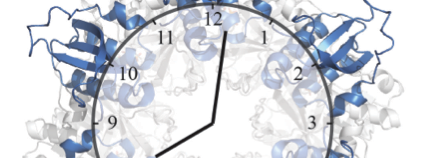

Almost all living things have adapted to earth’s day and night rhythm by keeping time with a biological clock. The biological clock of cyanobacteria, known as the Kai system, is one of the simplest known to date, and is therefore intensively studied by theoretical biologists and systems biologists in the hope to learn about biological time keeping. The Kai system is so robust, that it can even be reconstituted in the lab, simply by mixing the three Kai proteins (KaiA, KaiB and KaiC) in a test-tube. This test-tube oscillator still produces the same 24 hour rhythm that it does in the context of a living cell, and it can do so for weeks on end. The three Kai proteins interact with each other to produce these 24 hour rhythms, forming large protein complexes whose structure and composition changes depending on the time of day. The interactions between KaiC and KaiB marks a defining moment in the 24 hour cycle of the clock, but structural details of this interaction have not been well understood.
In the Proceedings of the National Academy of Sciences USA, Joost Snijder, Rebecca Burnley and Albert Heck, from the Biomolecular Mass Spectrometry and Proteomics Group (Utrecht University), have used structural mass spectrometry (native MS, ion mobility MS and HDX-MS) to shed light on the interaction between KaiB and KaiC. Their experiments revealed the composition of the KaiC-KaiB complex and showed which regions of the proteins are important for their interaction. Understanding these aspects of the KaiB-KaiC interaction explains many of the known features of the biological clock and helps us understand how such a simple system by biological standards is able to constitute a complex time keeping device in cyanobacteria.
The work was performed in collaboration with the groups of Alexandre Bonvin (Computational Structural Biology, Bijvoet Centre, Utrecht University) and Ilka Axmann (Institute for Theoretical Biology, Universitätsmedizin Berlin)
Link to the full article:
http://www.pnas.org/content/111/4/1379.long

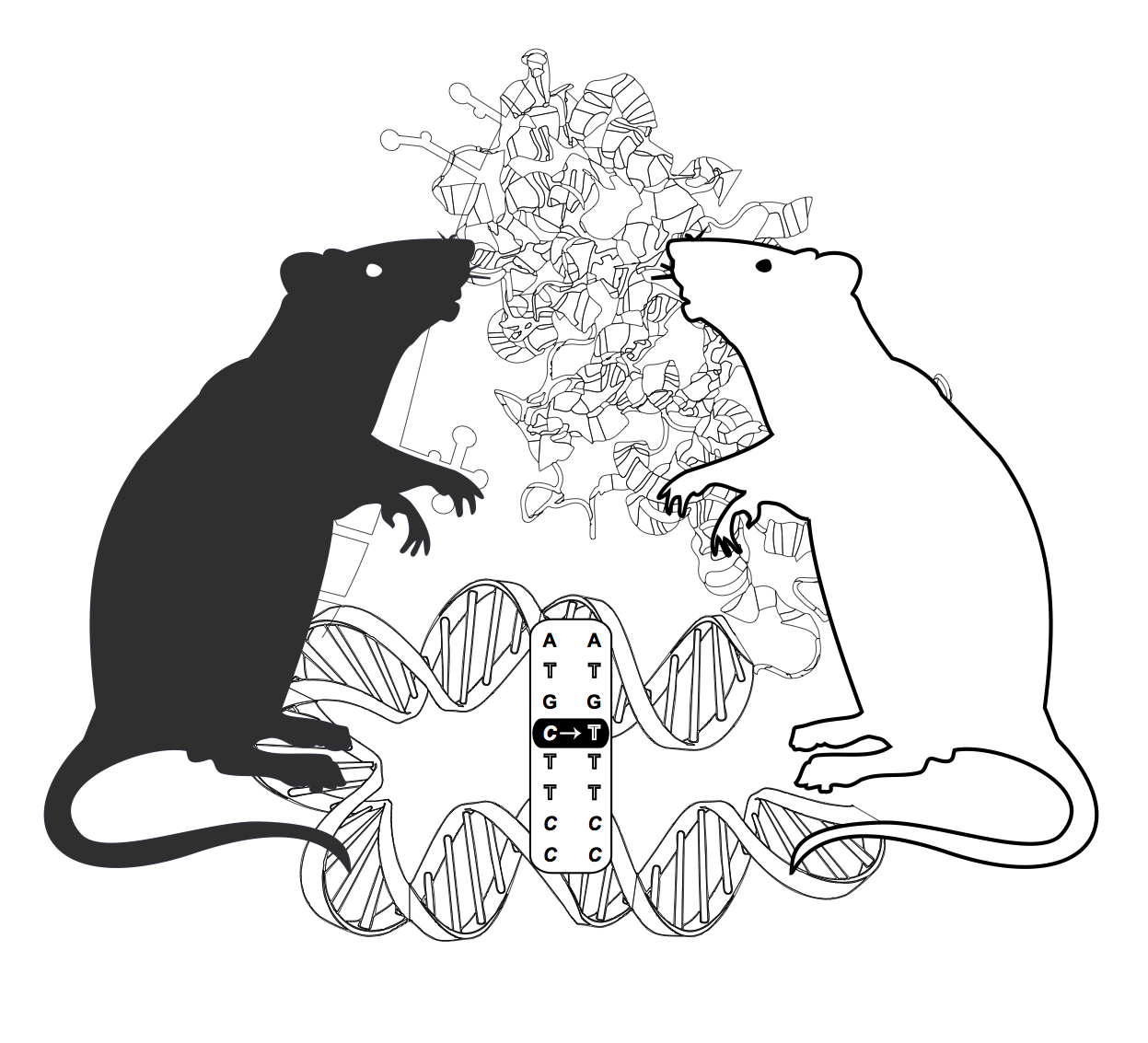
Experts in proteomics from Utrecht University and next-generation DNA sequencing from the Hubrecht Institute/UMC Utrecht collaborated in a study to systematically determine the consequences of genetic variation on the transcriptome and proteome. The researchers applied ultra-deep quantitative proteomics, whole genome sequencing and RNA-seq to liver tissues from different inbred rat strains. One of these inbred rats, the spontaneously hypertensive rat (SHR), is a widely used disease model for hypertension.
Proteomics experiments are normally hampered by identification methods based on reference genomes. Therefore, the authors designed a personalized protein database for each rat strain examined. They did so by integrating small genomic variants detected by whole genome sequencing and novel splicing and editing variants detected by RNA-seq. This extra information, in combination with the use of 5 different proteases, to extend the proteome coverage, resulted in the largest proteome to date (~13,000 proteins), with over 30% more proteins identified in a single sample than the current standard. Also, hundreds of novel genes, editing sites and transcript isoforms were identified at the protein level for the first time.
Besides this impressive gain in protein identifications subsequent integrated quantitative RNA and protein comparisons provided interesting novel insights in disease biology. Four differentially expressed genes popped up that had previously been associated with hypertension. One of those genes, Cyp17a1, was previously identified as one of the top hits in human hypertension GWAS studies.
 The identified link to hypertension is an illustrative example on how integrative genomics approaches can be used to dissect disease genes and mechanisms.
The identified link to hypertension is an illustrative example on how integrative genomics approaches can be used to dissect disease genes and mechanisms.
‘We have pushed the limits of current genomics and proteomics technologies’ says Prof Edwin Cuppen from the Hubrecht Institute and University Medical Center in Utrecht and one of the corresponding authors on the article. ‘Both in terms of the number of identified proteins as well as their characteristics at the RNA and protein. We demonstrate that the used techniques are now mature and can be used for routine integrative biology approaches to study effects of genetic variation on protein characteristics’
Prof Albert Heck from Utrecht University adds: ‘Our work shows that one plus one is more than two and that additional and relevant molecular details emerge when state-of-the-art technologies are combined. We predict our work to be a milestone, highlighting how next generation proteomics will be executed and can deliver new insights into disease biology.’
Link to the article on Cell Reports: http://www.cell.com/cell-reports/fulltext/S2211-1247(13)00640-2
Take a moment to view this brief Dutch film about NPC scientific director Albert Heck and his research group (Utrecht University)!
http://www.youtube.com/watch?v=Zm5-aKzezow

Next-generation proteomics: towards an integrative view of proteome dynamics
The proteome is extremely multifaceted due to splicing and protein modifications, further amplified by the interconnectivity of proteins into complexes and signaling networks that are highly divergent in time and space. Proteome analysis heavily relies on mass spectrometry (MS). MS-based proteomics is starting to mature enabling for the first time a near complete proteome coverage, including important post-translational modifications, of a given cell or tissue. This emerging next-generation of proteomics has been reached through a combination of developments in instrumentation, sample preparation and computational analysis. In a recent review in the high-impact “genetics” journal Nature Review Genetics Altelaar, Munoz and Heck describe this progress highlighting recent applications.
Reference
Next-generation proteomics: towards an integrative view of proteome dynamics
A.F. Maarten Altelaar, Javier Munoz and Albert J.R. Heck
Nat Rev Genet. 14 (2013) 35-48.
In February this year our group participated in a promotional video clip for Dell in which they present their recently obtained data storage cluster for proteomics data.
When a fluorescent sea anemone glows on a coral reef, it’s using a protein. When we move, proteins help our muscles contract. And when we curse our wrinkles as we age, it’s the lack of the protein collagen we lament.
{youtube}tRfN5Os79P4{/youtube}
Proteins also tell us about human health. By identifying proteins linked with specific diseases, we can develop novel diagnostics and therapies. At Utrecht University in the Netherlands, the Mass Spectrometry and Proteomics Group is exploring new methods of protein research. They can identify a protein or peptide every 100 milliseconds, producing vast amounts of data that must be stored safely and easily retrieved. Often, of all the proteins defined for each project, a tiny number are significant. Quick access to data is crucial for efficient analysis and isolation of the proteins that can fuel drug discovery.
The group wanted a new storage solution that would prevent bottlenecks and cut out painstaking backups. The Dell DX Object Storage Platform emerged as the best fit, based on its simplicity, flexibility and integrated data protection. Instead of slow tape backups, the IT team now relies on mirroring and automatic replication. When the group looks after data for other organisations, it knows it’ll be safe and easy to access. And, crucially, researchers can download files twice as quickly because the platform has higher throughput. Without delayed downloads, all their time is dedicated to the discovery of patterns and anomalies in protein behaviour that can change the way we manage disease.
Watch the video to hear the Utrecht University team talk about the challenges and rewards of proteomics.