Reducing the parasite population with a virus
The hitherto fairly unknown Triatoma virus, which kills insects that spread the tropical disease known as Chagas disease, could be used as a pesticide to control the insect plague and thus the spread of the disease. Researchers of the Biomolecular Mass Spectrometry and Proteomics Group in collaboration with researchers of VU University Amsterdam have studied every little detail of the virus and published their findings on 28 April in Nature Chemistry.
According to assessments of the World Health Organisation, ten million people had Chagas disease in 2012. At least 20,000 people die from the disease every year. This tropical disease is transferred by parasites in blood-sucking insects. The disease can be treated in the first few months, but when a longer period of time has elapsed, patients can no longer be saved. The disease is prevalent in Latin America, but has meanwhile spread to the north and infections have been found in the North-American state of Florida.
Fighting the cause
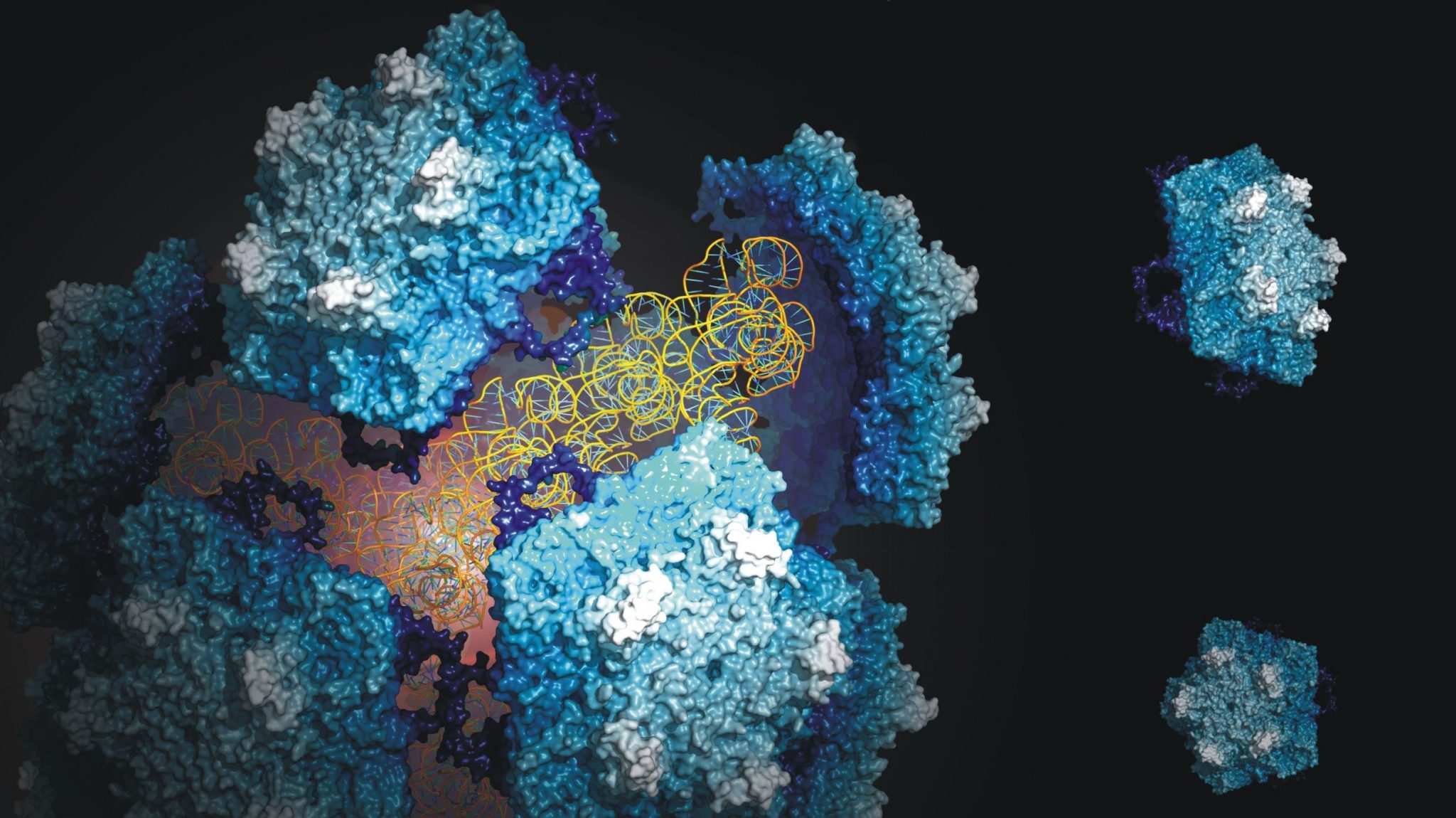
One of the best ways to prevent the spread of the disease is to combat the insect population that carries the pathogenic parasite. The Triatoma virus offers options in this respect, because infection with the virus kills the insects. In an attempt to chart this virus, researchers of the group of Professor Albert Heck (Utrecht University) in cooperation with the group of Dr Wouter Roos (VU University Amsterdam) have studied the composition, structure and stability of the virus in detail with the help of mass spectrometry and atomic force microscopy.
A complex collaboration
The researchers have shown that the stability of the virus is determined by a complex collaboration between the virus and its genetic material, which is strongly influenced by environmental factors. The experiments also provided information about the structure of the virus and the functioning of the genome. Both processes are vital to the virus’ life cycle. Furthermore, the data contributes to the design of viral systems in nano-technology and medicine.
Structure and stability
Albert Heck: “Our work has provided a lot of insight into the structure and stability of the virus. We have used many techniques in order to find out how to extract the genome from the virus and to reassemble the virus afterwards. This offers opportunities to add new elements to the virus which are potentially lethal to the host.”
This research was sponsored by the Netherlands Proteomics Centre, the Netherlands Organisation for Scientific Research (NWO) and the Foundation for Fundamental Research on Matter (FOM)
Publication
J. Snijder, C. Uetrecht, R.J. Rose, R. Sanchez-Eugenia, G.A. Marti, J. Agirre, D.M.A. Guerin, G.J.L. Wuite, A.J.R. Heck, W.H. Roos
Probing the biophysical interplay between a viral genome and its capsid.
Nature Chemistry